Frequently Asked Questions
What is the difference between refine.bio-processed and submitter-processed datasets?
Samples and the datasets they comprise are designated as refine.bio-processed if we were able to obtain raw data in a suitable format for one of our processing pipelines. If no suitable raw data is available for a sample on a supported platform, we obtain the processed data available from the source repository and modify it to be more consistent with refine.bio-processed data; these samples are termed submitter-processed. See the Source Data section for more information.
How do you process the data?
We process samples run on supported microarray platforms with Single-Channel Array Normalization (SCAN) and transcriptomic samples run on supported sequencing platforms with Salmon and tximport. Please see the Processing Information section for more details.
What type of data does refine.bio support?
refine.bio currently supports gene expression data, specifically genome-scale microarray and RNA-seq data. See our supported microarray and RNA-seq platforms.
What does “corrected” metadata mean?
Scientists will often use different terminology to refer to a similar sample metadata field or key.
For example, treatment and treatment protocol may make reference to the same kinds of information.
We attempt to perform some mapping between keys to aid in searches.
See the refine.bio-harmonized Metadata section for more information, including a full list of the mappings we perform.
Why do the values differ a little bit if I download different datasets?
We’ve prioritized keeping expression values consistent within a dataset based on the samples it contains. Specifically, we remove any genes that are not measured in every sample in a dataset and we do that prior to performing quantile normalization. When using quantile normalization, the expression value a gene is assigned in a particular sample depends on the rank of that gene. If a user download different datasets, which may have different numbers of genes, it’s possible then that the same gene in the same sample would have a different expression value between them.
Why do I get a limited number of genes back when I aggregate samples from different experiments?
When aggregating samples, we retain only the genes present in every sample. Different microarray platforms will often measure different sets of genes. RNA-seq samples in refine.bio can be quantified using different transcriptome indices, which may be built using different Ensembl releases and therefore include different sets of genes. Thus, when aggregating samples from multiple platforms, it is not uncommon for some genes measured in individual experiments to be dropped. The differences between gene sets included or measured for individual samples can be particularly pronounced when comparing older microarray platforms to more recent platforms. If the dataset delivered to you has fewer genes than you were expecting for that organism, it could be the result of combining multiple platforms (or the experiment may be from an older microarray platform).
Why can’t I add certain samples to my dataset?
refine.bio will sometimes obtain the metadata (e.g., sample title or experimental protocol) associated with a sample but the raw or submitter processed expression data files are in a format that we can not process. We do not allow you to add these samples to your dataset because we can not deliver expression values.
Why do the genes included in RNA-seq experiments change between experiments from the same organism?
You may find that a small proportion of genes are not consistently included in RNA-seq expression matrices from the same organism when you download data from refine.bio. The difference in gene sets can be detected when downloading multiple experiments as part of separate dataset downloads or when aggregating by experiment and comparing the genes included in expression matrices. (When aggregating by species to create a matrix comprised of samples from different experiments, genes that are not present in all samples are dropped.) When this occurs, it is because the experiments were quantified with Salmon transcriptome indices using different Ensembl releases of the same genome build.
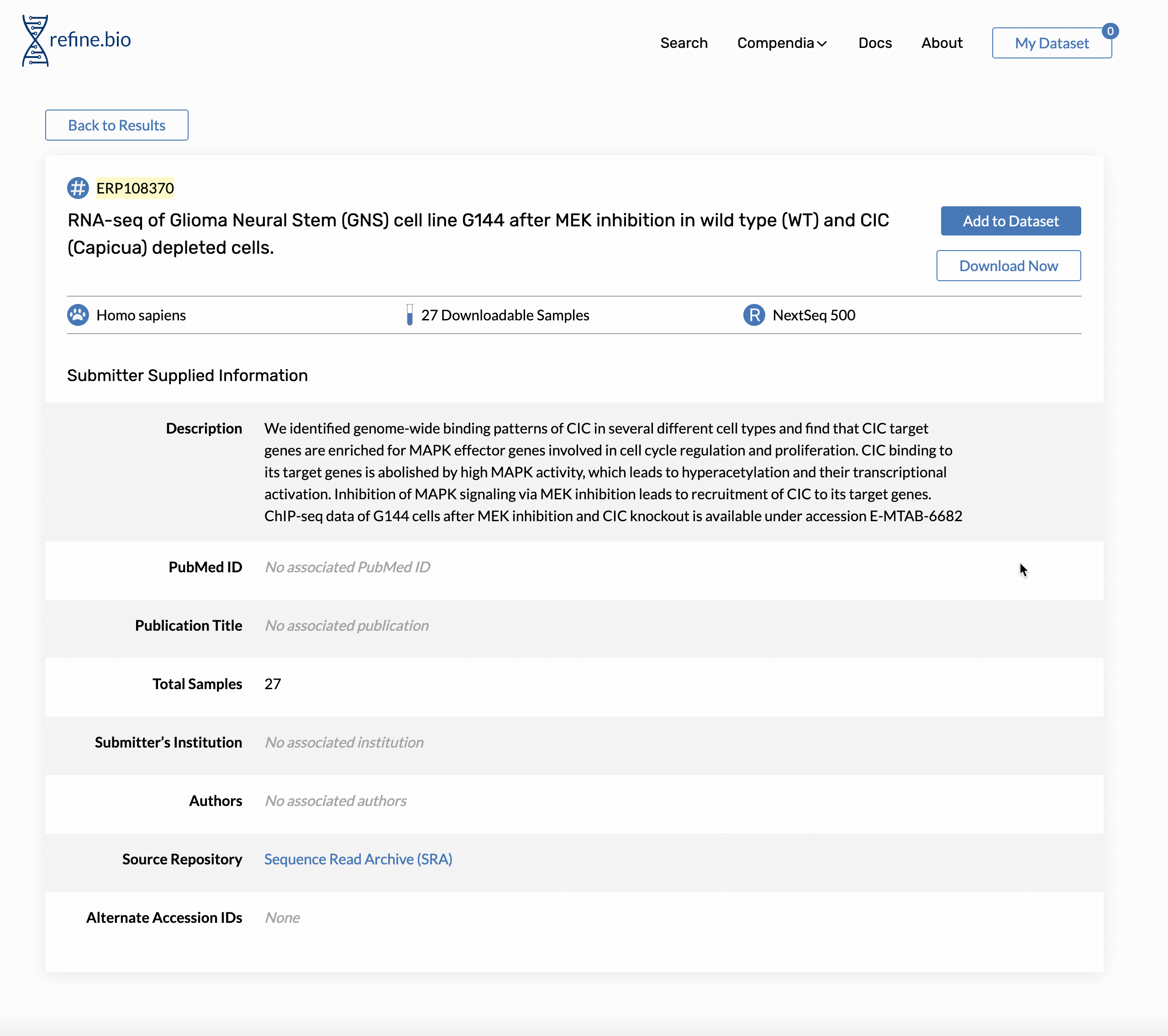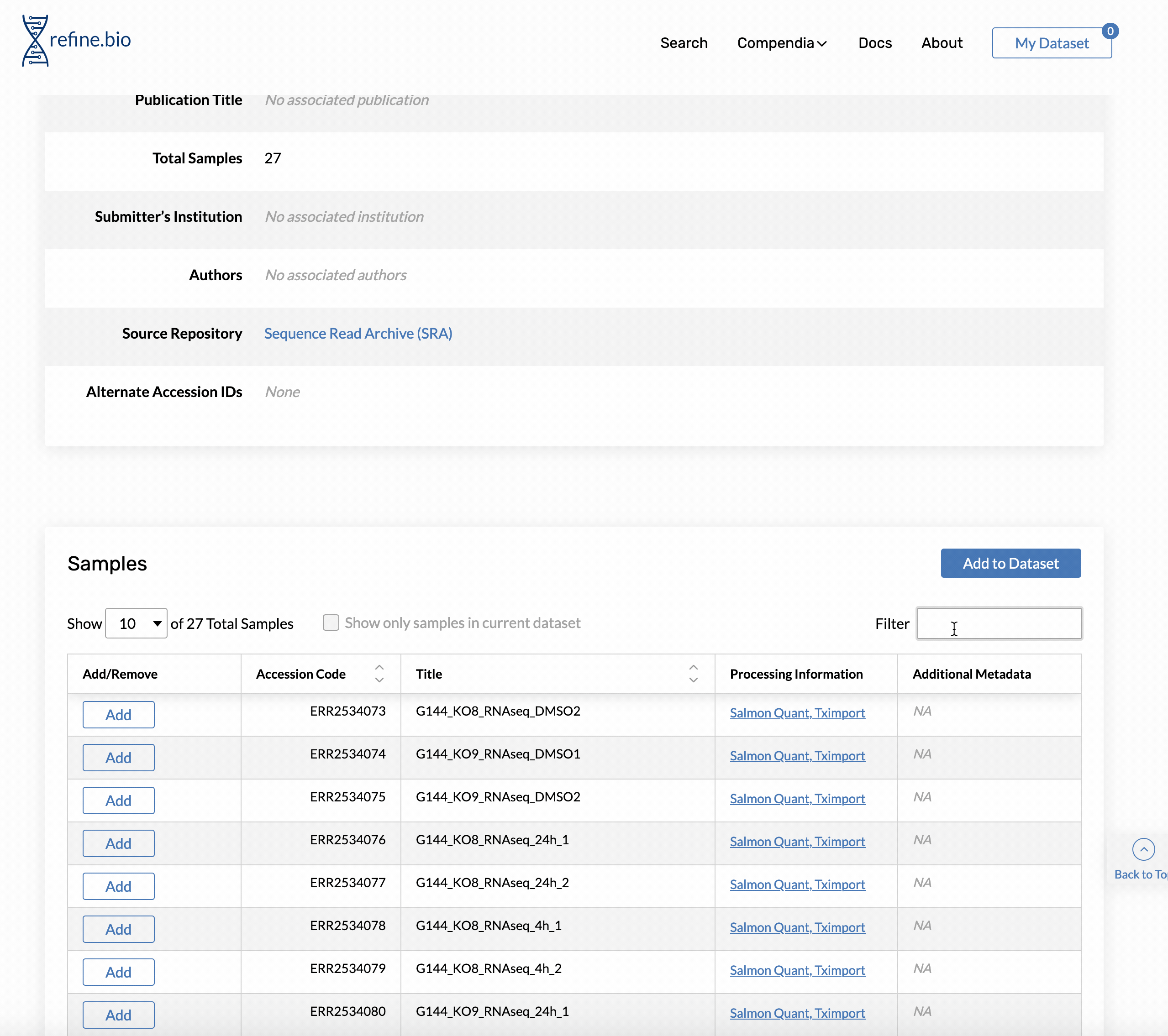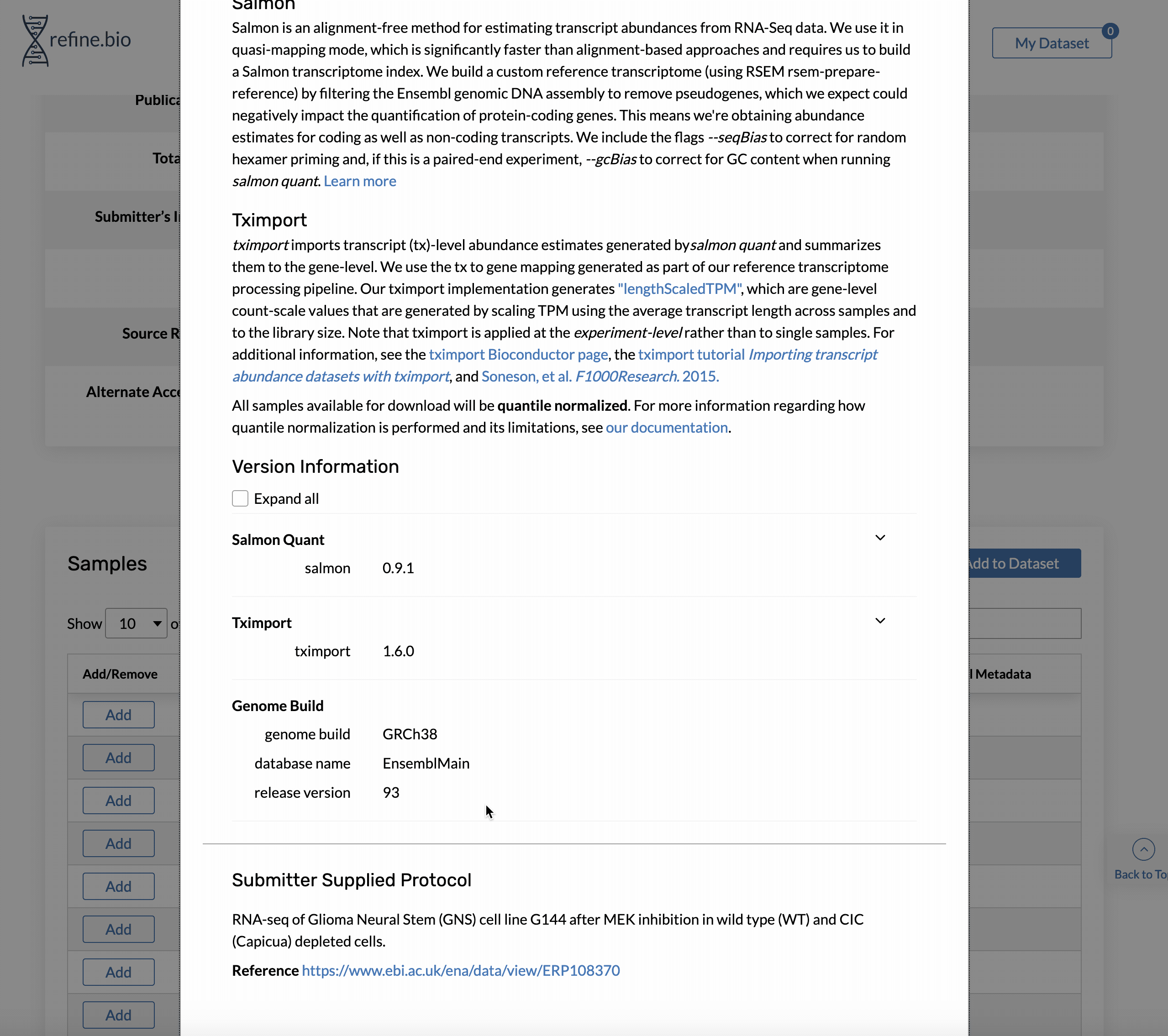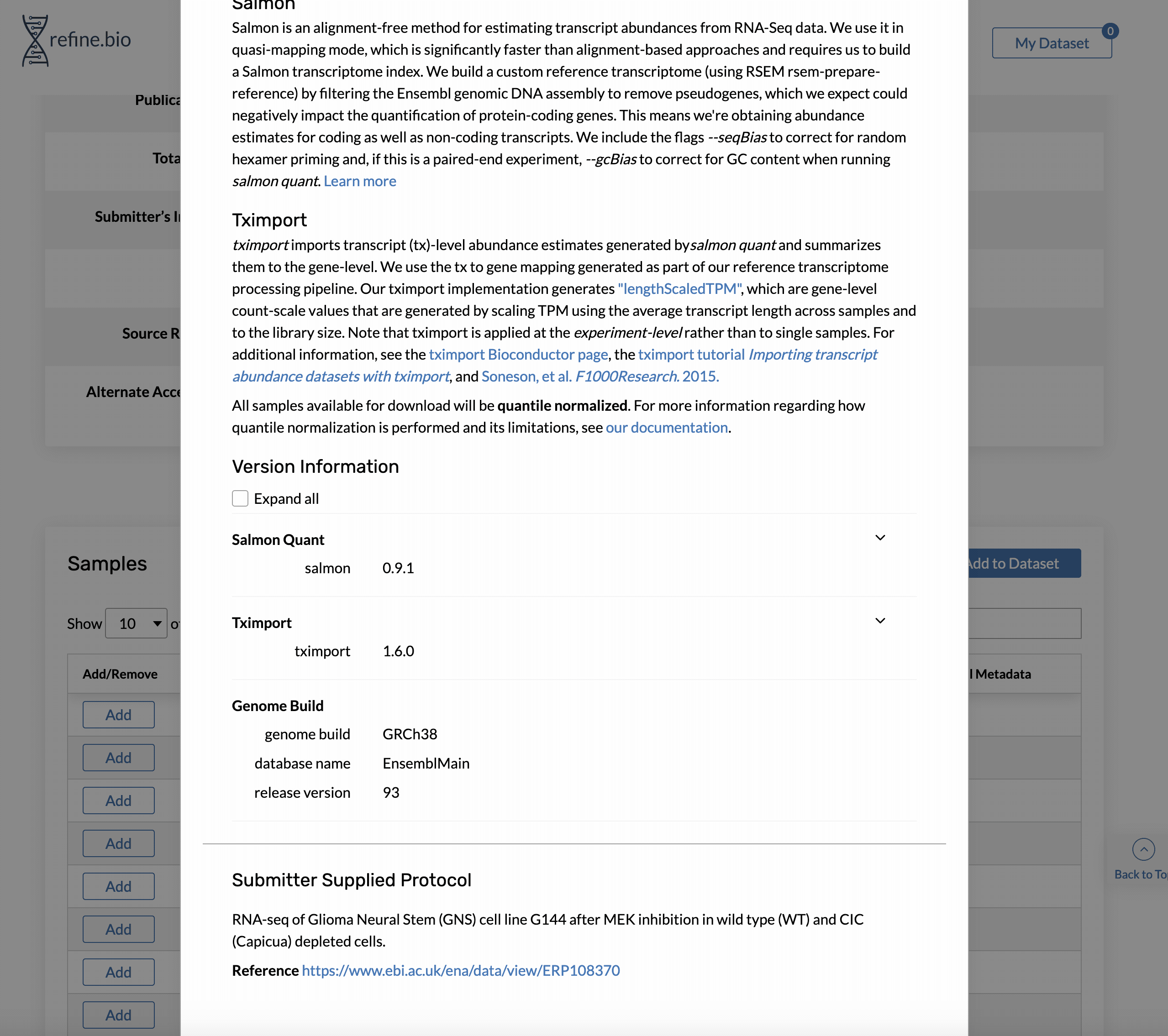
How can I find out what genome build and release were used to process RNA-seq data?
Genome build and Ensembl release version information is available on the pop-up displayed when you click a sample’s processing information link in the sample table on the page for an experiment.
The same information is available via our API. For the sample shown above, we could access the information with:
https://api.refine.bio/v1/samples/?accession_codes=ERR2534073
How can I find out what versions of software/packages were used to process the data?
Version information for the packages we think are most important for data processing is available on the pop-up displayed when you click a sample’s processing information link.

The same package information is in the processor list available via our API:
https://api.refine.bio/v1/processors/
In addition, you may wish to obtain our Docker images (prefixed with dr_) which will allow you to access version information for every dependency.
Are refine.bio datasets I download batch corrected?
We apply quantile normalization to mitigate issues caused by differences in the underlying distributions of gene expression values in samples. This makes the gene expression values broadly comparable, but doesn’t explicitly correct for batch, dataset, or platform. If the scientific question and analysis methods require datasets to be batch corrected, users should first investigate the existence of batch effects using methods such as Principal Components Analysis. If the source dataset is associated with major sources of variability in the data, users may wish to use a meta-analysis framework considering each dataset independently or to apply a batch correction tool. It may be sufficient to include batch, dataset, or platform as covariates for certain analyses.
Why are the expression values different if I regenerate a dataset?
The Regenerate Files button triggers the creation of a new dataset using the same options and including the same samples as the original, expired dataset.
If there are any changes to the way we process or aggregate datasets (e.g., target quantiles are updated) between the initial dataset creation and regeneration, these will be reflected in the new, regenerated dataset and may result in different values.
Users should take this into account when managing datasets obtained from refine.bio and take steps to appropriately archive datasets they use for analysis.
What does it mean to skip quantile normalization for RNA-seq samples?
If you would like to perform differential gene expression analysis with RNA-seq data obtained from refine.bio, you may want to choose to skip quantile normalization, as many methods designed for this problem expect unnormalized counts. refine.bio will not provide unnormalized counts when you skip quantile normalization, but it will provide output that can be used for testing differential gene expression (see Skipping quantile normalization for RNA-seq experiments for links to the relevant vignette). Note that skipping this step will make a dataset less comparable to other data obtained from refine.bio, as quantile normalization ensures that each sample’s underlying distribution is the same (see Quantile normalizing samples for delivery).
How do I cite refine.bio?
Please use the following:
Casey S. Greene, Dongbo Hu, Richard W. W. Jones, Stephanie Liu, David S. Mejia, Rob Patro, Stephen R. Piccolo, Ariel Rodriguez Romero, Hirak Sarkar, Candace L. Savonen, Jaclyn N. Taroni, William E. Vauclain, Deepashree Venkatesh Prasad, Kurt G. Wheeler. refine.bio: a resource of uniformly processed publicly available gene expression datasets. URL: https://www.refine.bio
Note that the contributor list is in alphabetical order as we prepare a manuscript for submission.